
Research Projects
Cyclodextrin Inclusion Complexes
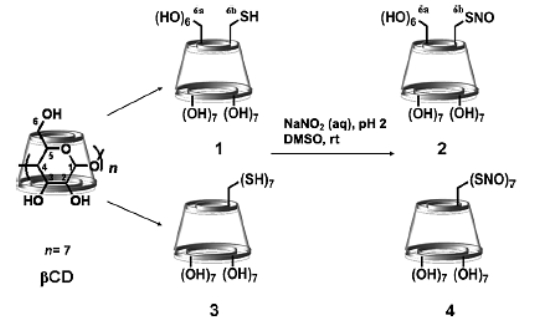
Synthesis of functional cyclodextrin derivatives
Structure of
macromolecules by X-ray
crystallography
A. Cyclodextrin Inclusion
Complexes:
crystalline state and solution structures and properties
Cyclodextrins are natural, water soluble macrocyclic carbohydrate
molecules
(Fig. A1), oligomers of
α-D-glucose,
that possess a cavity where a plethora of hydrophobic molecules can be
encapsulated. The resulting host-guest inclusion complexes exhibit many
characteristics of receptor-substrate systems. The laboratory has a long
experience in inclusion complexes of cyclodextrins with various guest
systems:
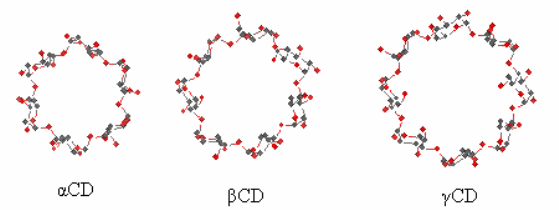
Figure A1. The common cyclodextrins: α-cyclodextrin (αCD), β-cyclodextrin (βCD) and γ-cyclodextrin (γCD).
Packing Research on X-ray structures of β-cyclodextrin inclusion complexes with model compounds has revealed that the majority of them forms dimers, which pack in 4 major packing arrangements [1-11]. The hydrophobicity / hydrophilicity of the end groups of the guest that emerge from the primary sides of the host dimer determine the crystal packing. A characteristic example is the difference in packing between the complexes of long aliphatic mono-carboxylic acids and di-carboxylic acids (Fig. A2).
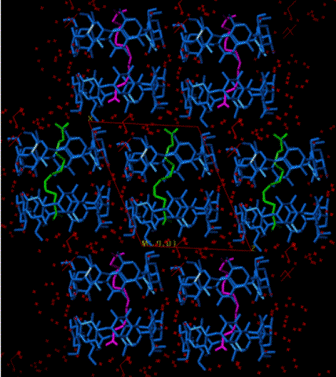
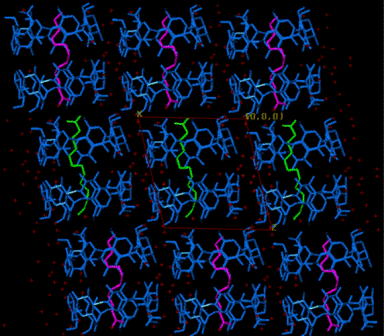
Figure A2. Inclusion of long mono-carboxylic acids (right, CH packing)
and di-carboxylic acids (left, Intermediate packing) into
βCD.
It is suggested
that it is the hydrophobic end-methyl group of the former guests at one
of the primary faces of the βCD
dimer,
what
dictates the channel formation in order to shield that part of the
guest from the surrounding water molecules. The carboxylic
end-group
of the guest is
found
entrapped inside
the channel at the other primary
face. However, two carboxylic groups
from
neighboring layers
self-associate
and form
carboxylic dimers, thus
stabilizing the whole system. In the
case of
dicarboxylic acid guests,
the
end-carboxylic groups
are exposed and
interact with water molecules.
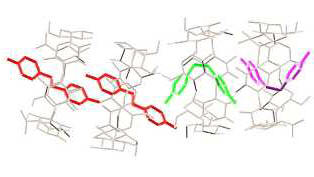
Of course, all rules have exceptions. Thus
flexibility on the part of the guest [1,2-bis(4-aminophenyl)ethane]
that acquires two different conformations in the cavity of the
βCD
dimer (Fig. A3), results in a packing arrangement different that the
four defined above, which however, can be transformed approximately to
one of them, the
screw channel mode of packing [12 and references
therein].
Another guest, 4-pyridinealdazine, that has the tendency to form π-π dimers on the one hand, and H-bonds with water molecules on the other, is encapsulated into βCD with the accompanying water molecules and forms βCD trimers (Fig. A4), detected for the first time [13], with host / guest ratio 3:2, a highly unusual stoichiometry.
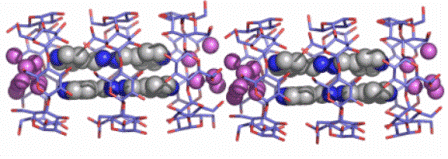
Fig. A4. Two βCD trimers forming channels. Each trimer holds a 4-pyridinealdazine dimer (guests in grey) and water clusters (pink) connecting by H-bonds the guests.
II. Self-assembly in solution and solid state
Non-bonding interactions, H-bonding and organization of CD
supramolecular systems in the crystalline state and in aqueous solution
have been extensively studied [14-18], as in
the following cases where the organisation is due to the guest
(Fig. A5 and A6)
.
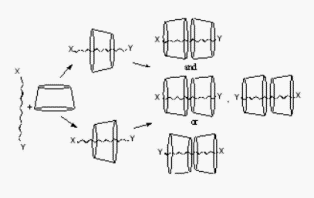
Fig. A5.
With
guests 1 - 4,
organization
of
cyclodextrins
in dimers
has
been detected in aqueous solutions [14].
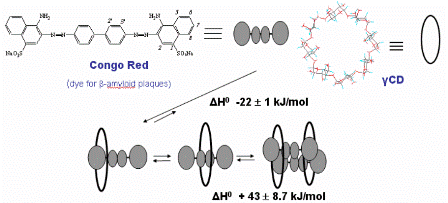
Fig A6. Exothermic rotaxanation of Congo red in
γCD
and endothermic dimerization of the complex [16].
On the other hand, anionic
cyclodextrins
(in the form of triethylammonium salts)
interact with the picrate salt of a positively charged CD to form stable
heterodimers in solution (Fig. A7). The association constants,
Ka, in DMSO-d6
and in DMSO-d6/H2O
(80/20, v/v) range from ~106 M-1 to ~104
M-1.
Multivalency in the interactions is
manifested by positive cooperativity,
negative enthalpy of formation
and sizeable negative entropy,
in support of development of well ordered
supramolecular structures in
solution
[19].
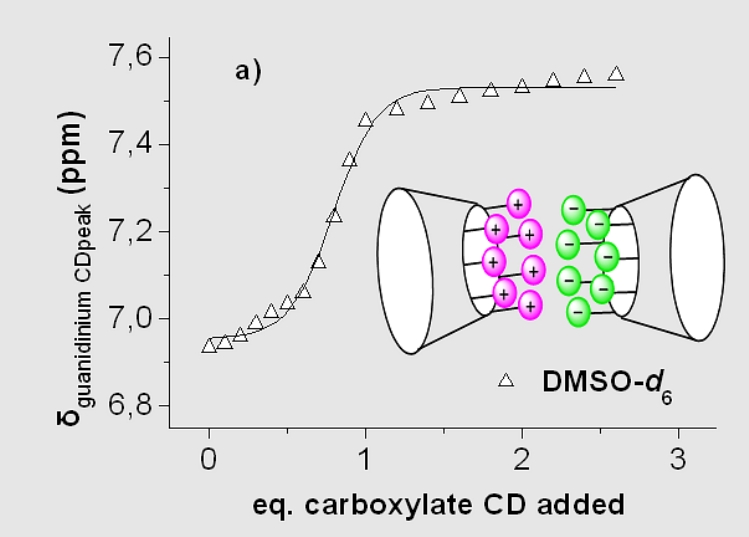
Figure A7.
Self-assembly of oppositely charged
CDs displaying positive cooperativity
III. Structural and recognition studies on inclusion complexes of cyclodextrins
Complexes with pheromones [20-27] and several plant growth factors [27,
28] have resulted in applications
for the
protection
the olive tree
by the
controlled release
of
pheromones
of the pests
Bactrocera
oleae and Prays
oleae
[29]. The
induced
fit
exhibited by the cavities of permethylated
α-
and
β-cyclodextrins
that resulted in the resolution of the racemic mixture of
Bactrocera oleae (Scheme A1 and Fig. A8)
is very remarkable. [30-31].
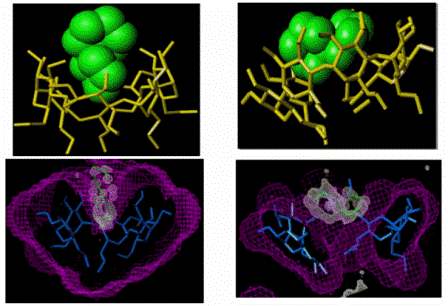
Fig. A8.
(Left):
Molecular
structure of the complex
of
permethylated
α-CD/(R)-spiroacetal
selectively precipitated from the racemic mixture of the guest. (Right):
Structure of the complex of permethylated
β-CD/(S)-spiroacetal,
selectively crystallised from the racemic mixture of the guest. Top:
Models of hosts and guests. Bottom: Differential electron density map of
the guests and van der Waals map of the hosts.
IV. Inclusion complexes with drug
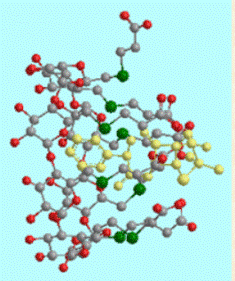
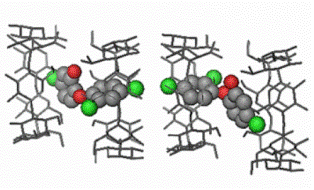
Fig A10. The inclusion complex of the
antibacterial agent
triclosan in
βCD
forms dimers packing in channels.
The
dichlorophenyl ring of the guest enter the
cavities of
βCD
dimers from their primary sides,
whereas the hydrophilic chlorophenol ring
extends in the space between dimers.
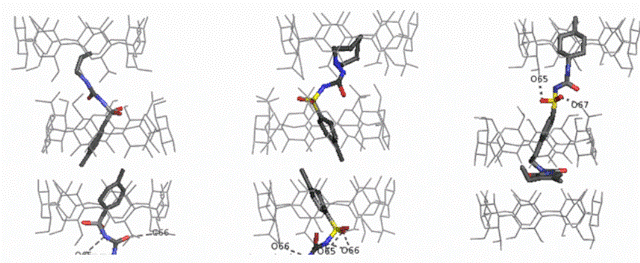
Fig A11. From Left to right the structures of the inclusion complexes of
the
hypoglycemic drugs
TBM, TLZ and GLP in
βCD.
The mode of inclusion of the drugs is the same:
the hydrophobic groups enter
the cavities of two hosts, belonging to adjacent
βCD
dimers from
the primary sides, whereas the hydrophilic sulfonylurea moiety is
located between dimers, forming H-bonds with the hosts. The packing of
the
TBM, TLZ complexes is in channel mode, whereas in that of the GLP the
βCD
dimers
are placed further apart (Intermediate mode) due to the increased length
of the guest.
Figure A12.
Binding of nucleotides to per(6-guanidino-6-deoxy)cyclodextrins
in aqueous solution
B.
Synthesis of
functional cyclodextrin derivatives
I.
Modified CDs display novel properties
Synthetic modifications of cyclodextrins result in introduction
of functional groups that endow the host molecules with specific
properties [1-3] ( Fig. B1), allow elongation of the host cavity to
capture long guest molecules or the combination of the above
that permits direct inclusion in specific direction [4,5] (Fig. B2).
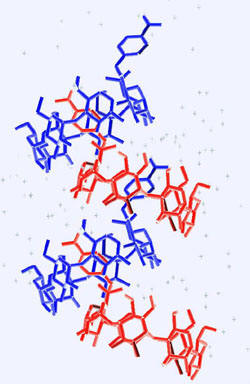
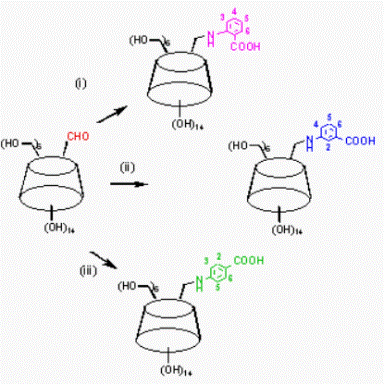
Fig.
B1. Synthesis of mono-aminobenzoic acid-substituted β-cyclodextrins and
supramolecular self inclusion in the crystalline state [1].
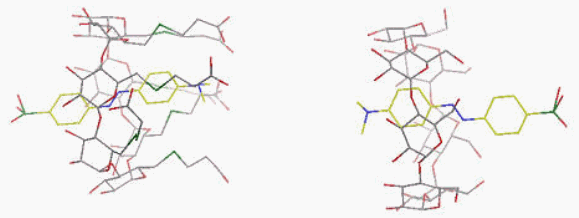
Fig.
B2. Per(carboxyethylthio)-β-cyclodextrin encapsulates the elongated dye
methyl orange (left) in a sense opposite to that of natural β-cyclodextrin
(right) [5].
Per-6-modified CDs with guanidino or lysine- or arginine-like groups (Fig. B3) comprises a very interesting family of hosts that have the ability to (i) penetrate cell membranes, (ii) transport molecules (iii) compact DNA and therefore perform transfection [6,7]. These properties have been used for transfection of green fluorescent protein (GFP) (Fig. B4) .
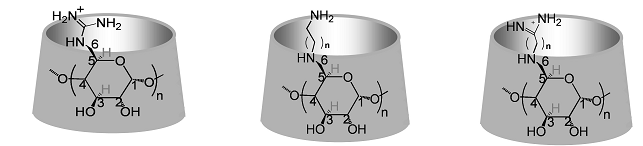

Fig B4. Guanidino and amino cyclodextrins:
(left)
penetrate cell membranes
(fluorescent microscopy image of HeLa
cells)
and (right)
perform DNA transfection that expresses GFP protein
(green fluorescence)
.
Fig. B5.
Positively
charged
β-
and γ-cyclodextrin: A Bacillus anthracis toxin pore blocker
III.
Negatively charded CDs with a range of applications
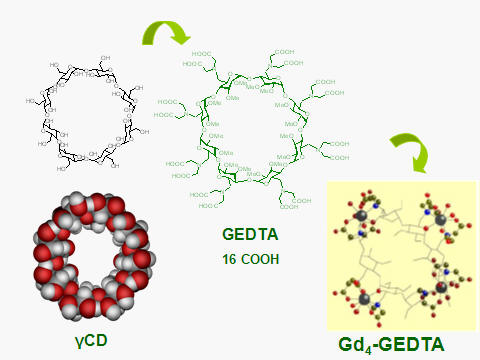
EDTA-type CDs, i.e. CDs modified with aminodiacetyl groups have been
synthesized, and characterized
(Fig. B6)
[10]. Their main property is the ability to
coordinate with metal ions, especially lanthanides and particularly
gadolinium, Gd(III).
The EDTA-CDs
coordinate with Gd(III) and fast exchanging water molecules to form
metal clusters that display high relaxivity values, especially at high
(100 MHz) magnetic fields.
A stable,
well-packed undecenyl-cyclodextrin (DMBUA) monolayer on Si/SiO2/ novolac
resin (AZ) substrate (Fig. B8)
is able to
detects triclosan from a 10 nM
aqueous solution in a reflectance
spectrometer [12].
Fig. B8. Representation of a stable CD monolayer able to detect the antibacterial triclosan (shown schematically).
Fig. B9. Representation of
the
supramolecular system conducting current between the Au substate and the
Pt/Ir
STM tip.
VI. Cyclodextrin derivatives for production of Reactive Oxygen Species (ROS) and Reactive Nitrogen Species (RNOS)
Thus

Fig. B10. A supramolecular nanoaggregate able to produce light-triggered
ROS and RNOS
C.
Structure of macromolecules by X-ray crystallography
X-ray crystallography allows to “look” at the detailed structure of biological macromolecules in three dimensions at the atomic level. This is essential in order to understand the macromolecules’ function (or malfunction in case of disease), to propose reaction mechanisms or design drugs. Current and past projects include:
I. Binding of γ-cyclodextrin to Glucogen Phosphorylase b
Selective binding of
γ-cyclodextrin
to Glucogen Phosphorylase b
(GPb) at
the glucogen storage site of the enzyme (Fig. C1) [1]. In order to
investigate whether the storage site can be exploited as target for
modulating hepatic glucose production,
α,
β
and
γ-cyclodextrins
(CDs) were identified as moderate mixed-type inhibitors of
GPb ( Ki
values 47.1, 14.1 and 7.4 mM for
α,
β
and
γ-CD,
respectively), the
γ-CD
being better than the others.
The structures of GPb with
β-
and
γ-CDs
provide an understanding of the binding, which is analogous to this of linear maltopentaose (G5) and maltoheptaose (G7) oligomers namely, anchoring of
two glucose residues to GPb. However, the inhibition constant of
γ-CD
is almost 7-times higher than that of G7 and of
β
–CD even higher. The lower potency of
γ-CD
compared to G7 is explained by lack of H-bonds between the
γ-CD residues
that are next to the anchoring residues and the protein, due to its
round shape.

Fig. C1. γCD (in red) binds to the storage site of the Glucose Phosphorylase b dimer.
II. Ferredoxins
Structural and electrochemical studies of the new family of 2[4Fe4S]
ferredoxins (Fds) from selected pathogenic bacteria
such as
Escherichia
coli (EcFd), Pseudomonas aeruginosa (PaFd),
Allochromatium
vinosum (AlvinFd,
Fig. C2) in order to correlate their structure to the widely different reduction potentials of their two metal clusters [–460
(cluster II) and –675 mV (cluster I)] [2,3].
The degree of exposure to the
outer environment of cluster I
sulfur atoms (Fig. C3)
leads towards less negative reduction potentials (E°). This is better
illustrated by V13G AlvinFd (high exposure, E° =
-594
mV) and EcFd (low exposure, E° =
-675
mV).
In
C57A AlvinFd
(Fig.
C3a),
the movement of the protein backbone, as a result of replacing the
non-coordinating Cys57 by
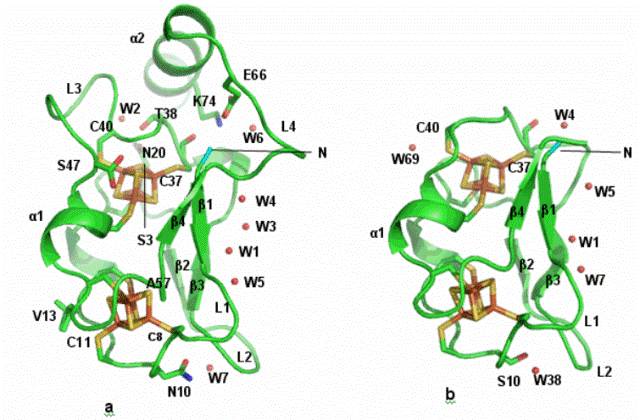
Fig. C2. The structure of the Alvin Fd variant, C57A (1.05 Å resolution, a). The environment of cluster II, although buried inside the molecule, is comparable to the exposed cluster II of the widely studied clostridia ferredoxin (b) that has two isopotential clusters (ca. –400 mV) . This is due to a water molecule (W6) entrapped in the interior of the C57A that attracts side chains of polar residues close to the cluster II S-atoms. In contrast, cluster I of C57A is less polar than cluster II of AlvinFd and of clostridia ferredoxins.
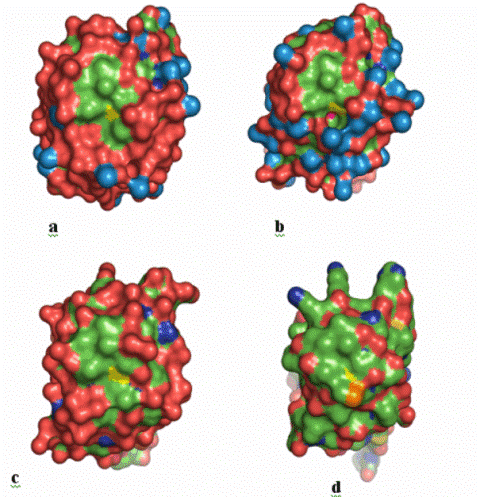
Fig.
C3.
Solvent-accessibility surfaces color-mapped
according to the chemical nature of the surface atoms of a, C57A; b, V13G;
c, PaFd; d, EcFd: Nitrogen, oxygen, and carbon atoms are shown in blue,
red, and green, respectively. The Cys11SG atoms of cluster I are shown
in yellow at the central region of the surfaces. The exposed S1 atom of
cluster I of V13G (b) is shown in magenta. The bulky sulfur atom of
Met13 of EcFd (d), in orange, limits the exposure of the Cys11SG atom.
III. Muscle proteins
Collaboration with the group of M. Wilmanns, EMBL-Hamburg on two muscle proteins titin and myomesin
1. The structure of the amino terminus titin/telethonin complex [4]: Titin (the largest protein made by human cells) along with myosin and actin, plays a crucial role in the muscle function. The structure of the 2:1 titin:telethonin complex gives the details of the interaction of two titin molecules with telethonin, a small protein-component of the the Z-disc of muscles (Fig. C4). This leads to a model of titin interaction in the Z-disc: two titin molecules from different sarcomeric filaments cross-linked at their amino terminus via telethonin. The study may lead to new insight for some muscle and cardiac diseases; moreover, it may provide a molecular paradigm about the cross-linking of major sarcomeric filaments.
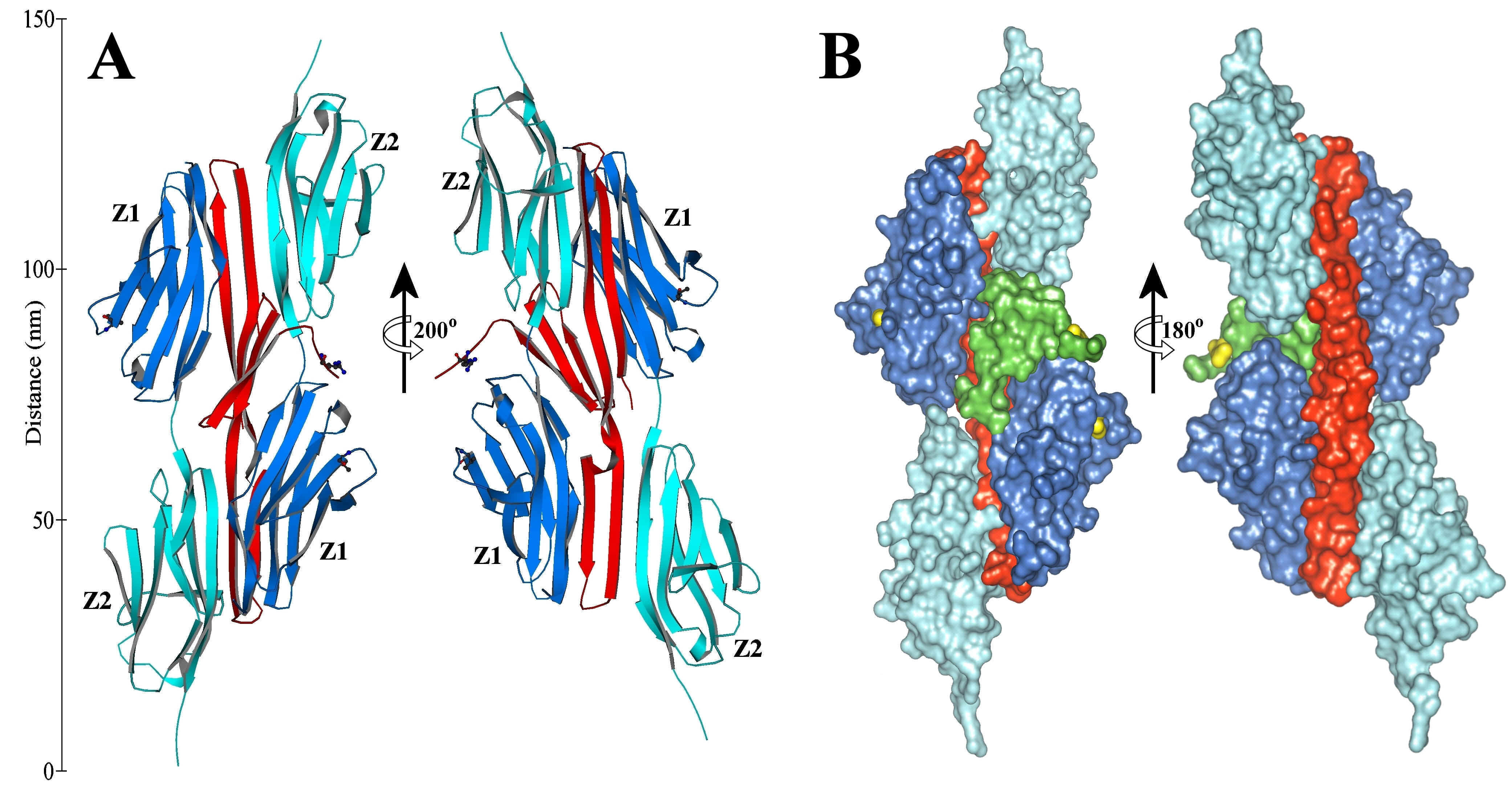
Fig. C4. A: ribbon representation (in two orientations) of the two antiparallel titin immunoglobulin-like domains Z1 (blue, residues 1-98) and Z2 (cyan, residues 99-196 including the Z1-Z2 linker) cross-linked by telethonin (red) via extended antiparallel β-sheets involving the three molecules. B: surface representation of the titin-telethonin-titin complex, in two orientations (in green, the telethonin domain 60-90 not participating in β-sheets).
2. The myomesin structure. The mechanical forces exerted by the muscles are sensed and buffered by unusually long and elastic filament proteins with highly repetitive domain structures. Myomesin is one such repetitive filament protein, which is thought to form bridges between the main contractile filament, that provides the muscle with resistance in the radial dimension. To investigate how the repetitive structure of myomesin contributes to muscle elasticity, the overall architecture has been determined using a combination of four complementary structural biology methods [5]. The structure of the myomesin domains my9-my11 has been elucidated by X-ray crystallography. Each domain comprises a immunoglobulin-like and helix pattern (Fig. C5 in green). These have been combined with the already available structure of the final my12-my13 domains of the carboxyl terminus in order to make a model of how the myomesin filament attaches in the M-band of the muscle sarcomer.
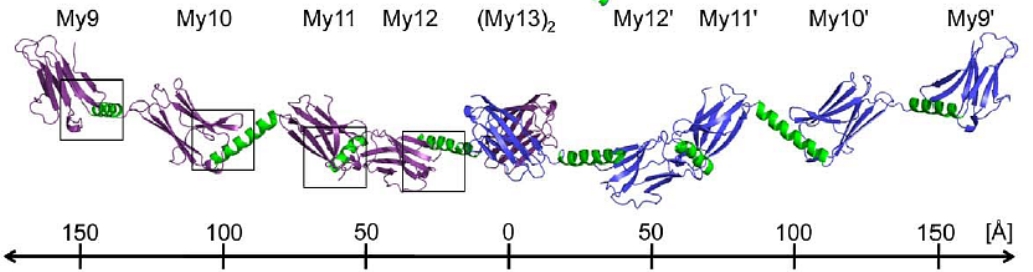
Fig. C5. The complete myomesin (My) tail-to-tail filament structure: In ribbon representation the dimeric myomesin IgH domain array My9–My10–My11–My12–(My13)2–My12
'–My11'– My10'–My9'. Myomesin domains that have been structurally investigated are shown in violet (first molecule) and blue (second molecule). The helical linkers are shown in green. A ruler provides an overall length estimate of the filament.IV. Endoplasmic Reticulum Aminopeptidases
Endoplasmic Reticulum (ER) aminopeptidases ERAP1 and ERAP2 cooperate to trim a vast variety of antigenic peptide precursors to generate mature epitopes for binding onto MHC class I molecules and help regulate the adaptive immune response. In collaboration with the group of E. Stratikos at our institution, we have determined for the 1st time the structure of ERAP2 to 3.08 Å by X-ray crystallography [6]. The ERAP2 structure (Fig. C6) provides a structural explanation for the different peptide N-terminus specificities between ERAP1 and ERAP2 and suggests that such differences extend throughout the whole peptide sequence. Overall, the structure helps explain how two homologous aminopeptidases cooperate to process a large variety of sequences, a key property to their biological role. Common coding single nucleotide polymorphisms (SNPs) in ERAP1 and ERAP2 have been linked with predisposition to human diseases ranging from viral and bacterial infections to autoimmunity and cancer. The common ERAP2 SNP rs2549782 that codes for amino acid variation N392K leads to alterations in both the activity and the specificity of the enzyme. Specifically, the 392N allele excises hydrophobic N-terminal residues from epitope precursors up to 165-fold faster compared to the 392K allele, although both alleles are very similar in excising positively charged N-terminal amino acids. This is primarily due to changes in the catalytic turnover rate (kcat) and not in the affinity for the substrate. X-ray crystallographic analysis of the ERAP2 392K (Fig. C6)allele suggests that the polymorphism interferes with the stabilization of the N-terminus of the peptide both directly and indirectly through interactions with key residues participating in catalysis [7]. The study provides mechanistic insight to the association of this ERAP2 polymorphism with disease and support the idea that polymorphic variation in antigen processing enzymes constitutes a component of immune response variability in humans.
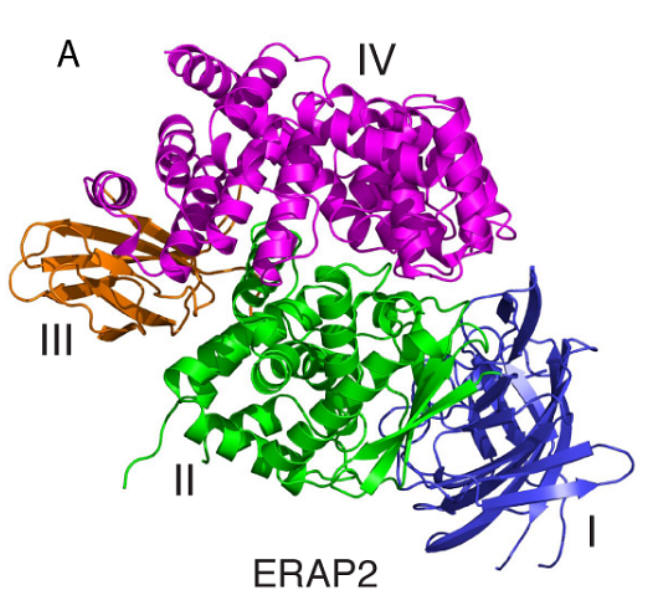
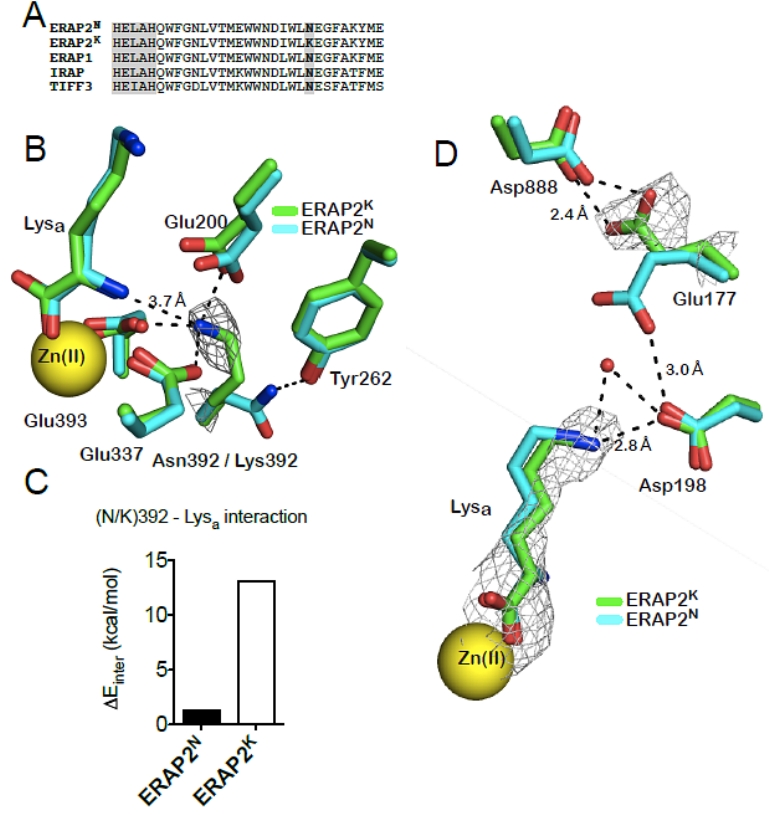
Fig. C6. Left: Ribbon representation of ERAP2 colored by domain (domain I in blue, II in green, III in orange and IV in magenta). Right: A, sequence alignment of ERAP2 and homologous aminopeptidases showing conservation of the polymorphic residue 392 (in bold); the adjacent aminopeptidase motif (HELAH) is also indicated; B, key catalytic residues in the ERAP2K structure are shown in stick representation and superimposed with equivalent residues in ERAP2N (2SE6). Note the relative positioning of Lys392 with respect to the catalytic Glu residues as well as the N-terminus of the bound ligand (Lysa). Electron density 2|Fo|-|Fc| at 2σ is indicated around Lys392; C, pairwise electrostatic interaction energies (ΔΕinter in Kcal/mol) calculated between the polymorphic residue 392 and the Lysa ligand. The reported values are the average of the 20 runs with a standard deviation of less than 1%, which represents the uncertainty due to the granularity of the grid; D, superimposition of ERAP2 residues that cap the S1 specificity pocket of the enzyme. |Fo|-|Fc| electron density (at 2.5σ) calculated in the absence of the ligand is shown around Lysa. 2|Fo|-|Fc| electron density at 2σ is indicated around Glu177.
V. RNA Complexes
Structure determination of complexes of
the ribosomal decoding aminoacyl site (A-site) of bacterial 16SrRNA
constructs with
synthetic analogs of aminoglycosides. Aminoglycoside
antibiotics selectively bind to the A-site of bacterial 16SrRNA,
interfering in the fidelity of proteosynthesis in the bacterial cells.
The designed compounds are synthesised at the collaborating
organic Chemical Biology
laboratory of our institution headed by Dr D. Vourloumis. Elucidation of
the structure of the
above compounds in complex with selective RNA constructs that
are appropriate models of the A-site, provides the prerequisite for
guiding this synthetic effort.
Moreover, the
structural information combined with biochemical and kinetic experiments
is the first step in the development of pharmaceuticals for fighting
bacterial infections by RNA-directed therapies.
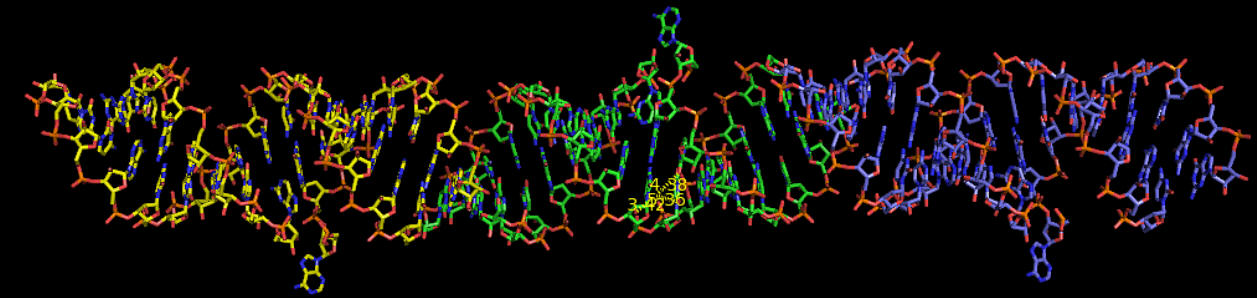
Fig. C7. Model of the structure of a ligand-free bacterial RNA construct. Three consecutive molecules of RNA monomers (each represented by a different colour) form infinite continuous chains. The structure has been solved by the software IL MILIONE (Burla MC, Caliandro R, Camalli M, Carrozzini B, Cascarano GL, De Caro L, Giacovazzo C, Polidori G, Siliqi, D, Spagna R, J Appl CRYSTALLOG. 2007, 40 609-613)
VI. DsbD: a bacterial thiol-disulfide oxidoreductase
DsbD is one of the five proteins of the bacterial Disulfide bond (Dsb) formation system. It is located in the inner membrane of Gram-negative bacteria and it is responsible for transporting reducing power from the cytoplasm to the oxidising periplasm of the cell (Fig. C8). It comprises three domains; the central transmembrane domain (tmDsbD), of unknown structure, is flanked by two globular periplasmic domains, the N-terminal domain nDsbD) with an immunoglobulin-like fold and the C-terminal domain (cDsbD) with a classical thioredoxin (Trx) fold. DsbD plays an important role in oxidative protein folding because it allows for the correct formation of disulfide bonds in proteins functioning in harsh extracytoplasmic environments [8]. It is also involved in bacterial pathogenesis as the expression and stability of most virulent factors (secreted molecules, secretion apparatuses, adhesion systems etc) are dependent on the presence of DsbD. The structures of the N-terminal domain in the reduced form and of two point-mutants of the C-terminal domain have been determined [9,10] in collaboration with the Department of Biochemistry, University of Oxford (Prof. Stuart J. Ferguson, Prof. Christina Redfield and Dr Despoina A.I. Mavridou). This work contributed significantly in elucidating the interaction of the soluble domains of this unique oxidoreductase but also in the general understanding of the factors controlling the reactivity of the ubiquitous thioredoxin fold.
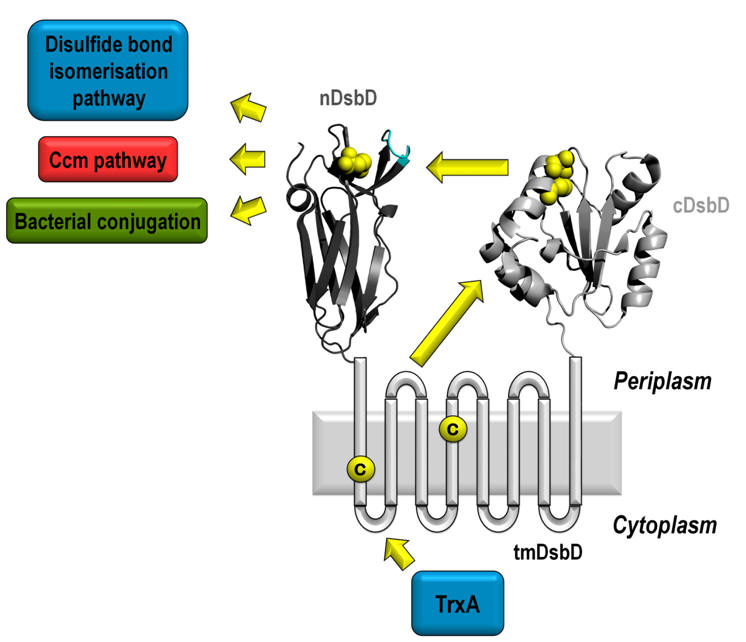
Fig. C8. DsbD: The sole reductant provider in the periplasm.
New crystallisation
methodology for biological macromolecules, in
collaboration with
1.
As Coordinators of the
Industry-Academia Partnerships and Pathways -
Marie Curie Project TOPCRYST, we developed the use of Dual Polarization
Interferometry (Fig. C9), pioneered by Farfield Scientific Ltd., to probe
crystallisation at its most crucial stages. This allows to predict the
outcome of crystallisation trials when they are still at their earliest
stages and thus to rationally design and direct such experiments in
order to lead them to well-diffracting crystals [11]. Research on other techniques for
effective a priori prediction of crystallisation conditions, such as the
use of Genetic Algorithms and of calorimetry, is also being actively
pursued [12].
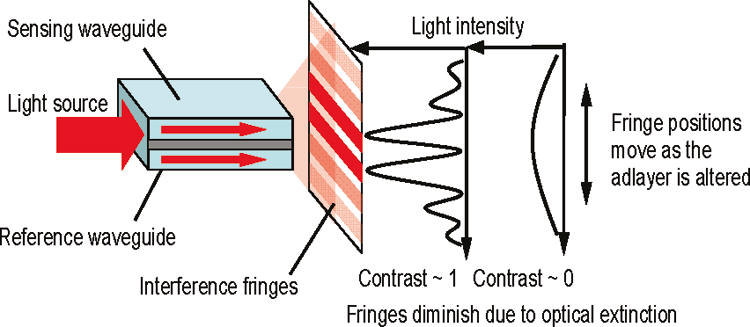
Fig. C9. Optical principle of dual polarization interferometry. Light from a sensing waveguide in contact with the investigated solution interferes with that from a reference waveguide, resulting in interference fringes characterized by their phase and contrast.
2. Research into substances and materials promoting the
heterogeneous nucleation of macromolecular crystals is a blossoming
topic in crystallogenesis. We have participated in developing the use of
materials containing pores of cavities, such as Bioglass, carbon
nanotubes, and Molecularly Imprinted Polymers (Fig. C10), as heterogeneous nucleants and are pursuing further ideas [13-16].
